Tik-61.181 Informaatiotekniikan erikoiskurssi I (4 ov)
Exercises 1 & 2 (Autumn 2000)
Jukka Lindström, 47798R
Palautettu 31.1.2001
Vastaukset tehtäviin
1. a), b), c), d), e), f), g), h), j), k), l), o)
2. a), b), c), d)
a) Explain in detail how you would align the following sequences with pair HMM:s and with non-probabilistic methods: AAAG and ACG (DEKM ch.2 and ch.4, S&M ch.3)
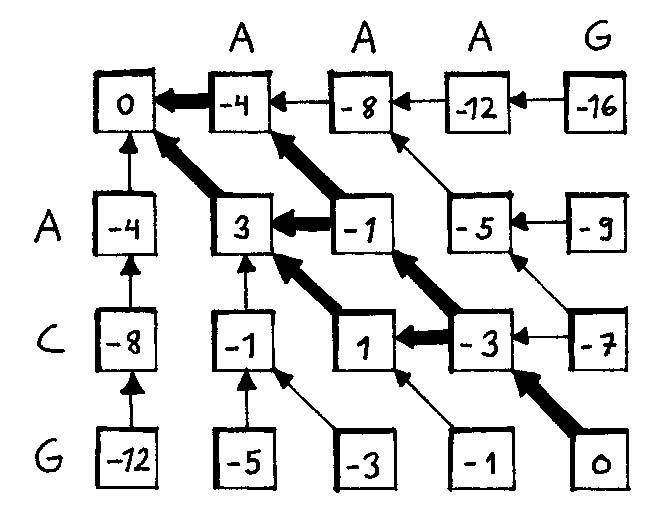
Tehtävässä pitää täsmätä sekvenssit ei-parametrisella metodilla ja käyttämällä Piileviä Markov -malleja. Ei-parametrisena menetelmänä olen käyttänyt dynaamista ohjelmointia globaalille tasaukselle (Needleman-Wunsch). Kuvassa on esitetty tasauksen onnistuminen. Parhaiten täsmäävä reitti etsitään takautuvasti, kun on löydetty suurin pistemäärä taulukon vasemmasta reunasta tai alareunasta. Tämä suurin arvo 0.
Pisteytysfunktiona käytin seuraavaa.
s(sama) = 3
s(eri) = -2
ja D:n eli tyhjän käyttämisen arvona on -4.
Kuvasta nähdään optimaalinen tasaus paksuimmilla viivoilla (vaihtoehtoiset yhtä hyvät tasaukset on katkoviivoilla). Eri tasausvaihtoehdoiksi saadaan seuraavat
AAAG AAAG AAAG
AC_G _ACG A_CG
Tässä on HMM-malli joka tunnistaa molemmat sekvenssit. Tätä pitäisi laajentaa pair HMM:ksi ja etsiä todennäköisyn reitti viterbi-algoritmilla, mutta jää väliin. (pitäisi löytää hyvä esimerkki tuon käyttämisestä, pelkkää algoritmia katselemalla homma ei oikein selvinnyt. Sen ymmärsin, että Viterbi-polun todennäköisyyden laskeminen ei ole kummoisempi homma kuin pisteytyksen laskeminen dynaamisessa laskennassa. Mutta kuinka päästä eteenpäin?
b)
Analyze briefly the relation of the probalistic approach and the
non-probabilistic approach for sequence alignment. (DEKM ch.2 and
ch.4, S&M ch.3)
Ei-probabilistiset menetelmät voidaan käsittää tilakoneina ja probabilistiset siirtymien todennäköisyyksillä laajennettuina tilakonena.
Ei-probabilistisissa menetelmissä käytetään tiloja vastaamaan merkkien lisäämistä ja sovittamista, kun taas probabilistissa menetelmissä tilat vastaavat lisättyjen merkkien todennäköisyysjakaumia.
Ei-probabilistissa malleissa paras tasaus pyritään löytämään minimoimalla sakko tietyllä estimoidulla sakkofunktiolla. Probabilistisissa malleissa tätä vastaavasti pyritään löytämään tasaus, joka on todennäköisin (maksimoidaan todennäköisyys).
Ei-probabilistisessa mallissa sakko muuttuu siirtymien kautta, jolloin sakkofunktiota sovelletaan tasauksessa oleviin merkkeihin. Sakko määräytyy merkkien erilaisuuden mukaan. Probabilistisessa mallissa siirtymät vaikuttavat kokonaistodennäköisyyteen. Siirtymien todennäköisyydet on laskettu näytejoukkojen perusteella.
Sakkofunktion hienojakoisuus vaikuttaa siihen kuinka monta eri optimaalista ratkaisua saadaan. Sakkofunktion valinta vaikuttaa täsmäykseen paljon. Todennäköisyysmallissa käytössä ollut näytejoukko on kriittinen tasausta onnistumiselle ja sille kuinka monta "todennäköisintä" tasausta menetelmä antaa.
c) Give an example of the possible uses of the MC-model and HMMs in bioinformatics. Outline briefly the main differences between the two models. (DEKM ch.3)
Näihin kahteen liittyy läheisesti seuraavat kysymykset:
Kuuluuko annettu lyhyt palanen genomista CpG -saarekkeeseen?
Onko annetussa pitkässä sekvenssissä CpG-saarekkeita?
Yleisemmin nämä kysymykset voidaan esittää seuraavaasti - kuuluuko tietty sekvenssi haluttuun joukkoon tai onko annetulla sekvenssillä haluttuja ominaisuuksia (jos se kuuluu haluttuun joukkoon)?
Markov ketjuissa tilojen transitioihin on liitetty todennäköisyydet ja tiloissa emittoidaan ainoastaan yhtä symbolia (deterministinen). Transitiotodennäköisyydet riippuvat ainoastaan tilasta, jossa ollaan. Markov ketjulla voidaa tehdä erottelua CpG-saarekkeiden ja muun alueen välillä annetulle näytteelle, mutta saarekkeiden etsitää pidemmästä sekvenssistä ei pystytä toteuttamaan.
HMM:ssä tilojen transitioihin sekä tiloissa emittoitaviin symboleihin on liitetty todennäköisyydet. Todennäköisyydet riippuvat ainoastaan tilasta, jossa ollaan. Koska emittoidusta symbolista ei voida päätellä missä tilassa ollaan on malli nimetty piileväksi (hidden). HMM:llä voidaan käyttää nimeämään tuntemattoman sekvenssin osia tai sekvenssien etsintään. HMMmiä voidaan käyttää myös sekvenssien ryhmittelyyn (mm. pairwise-HMM)
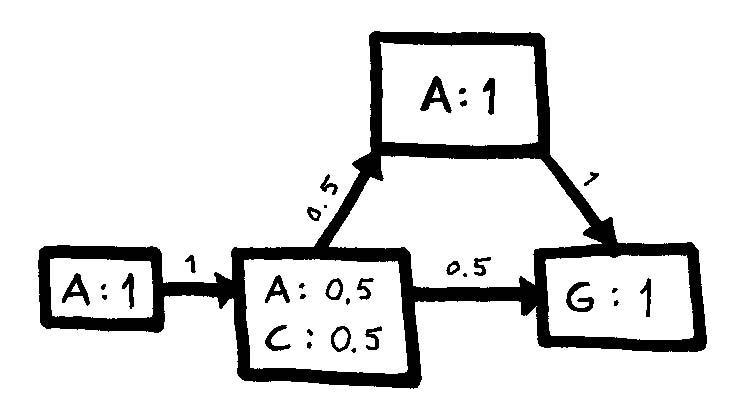
d) Draw the state model graph with transition probabilities and calculate the probability of seeing the output sequence { cola, lemonade } if the machine starts in the WP state. What kind of behaviour would make this HMM to a (visible) Markov model?
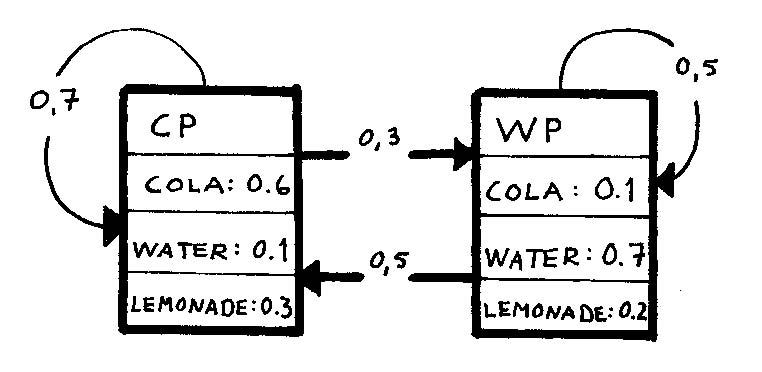
P({cola, lemonade}) = P(cola|WP) * ( P(->WP|WP) * P(lemonade|WP) + P(->CP|WP) * P(lemonade|CP) )
= 0.1 * (0.5 * 0.2 + 0.5 * 0.3) = 0.025
HMM:t ovat laajennus Markov-ketjuihin. Markov ketjuissa tiettyssä tilassa emittoidaan vain tiettyä vastetta ja todennäköisyydet liittyvät ainoastaan tilojen transitioihin. Markov ketjussa tiedetään aina mihin tilaan saatu observaatio kuuluu. HMM:ssä tietyssä tilassa voidaan eri todennäköisyyksien mukaisesti emittoida eri vasteita. Ylläolevasta mallista voitaisiin rakentaa helposti approximoiva Markov ketju (eli näkyvä Markovin malli) asettamalla suurimman todennäköisyyden symboli aina tietyn tilan emittoimaksi symboliksi (eli CP tilassa emittoitaisiin aina colaa ja WP:ssä aina vettä). Malliin voidaan liittää uusiakin tiloja.
e) Do excercise 3.3 in DEKM and explain its result briefly in the bioinformatics framework.
Tehtävästä
3.2 saadaan tulos, että kaikkien L pituisten sekvenssien
todennäköisyyksien summa on
![]() .
Tästä saadaan helposti todistettua geometrisen summan
kaavan avulla:
.
Tästä saadaan helposti todistettua geometrisen summan
kaavan avulla:
![]()
Todennäköisyydet mallille on saatu jostain tietystä rajoitetusta näyteaineistosta. Ylläolevan mukaan Markov ketju -malli kuitenkin pystyy kuitenkin arvioimaan todennäköisyydet kaikille eri pituisille sekvensseille.
f) Compare briefly the FSAs and pairwise HMMs in searching. (DEKM ch.4)
Tähän pätee pitkälti samat asiat kuin kohdassa b) sillä FSA on deterministinen menetelmä ja pair-HMM on probabilistinen menetelmä.
FSAssa käytetään tiloja vastaamaan merkkien lisäämistä ja merkkijonon sovittamista. Siirtymiin liittyy sakkofunktio symbolien erilaisuuden mukaan. Pyritään löytämään se tasaus, jolla on pienin sakko. Menetelmät toimivat kirjan mukaan erittäin hyvin, koska parametrit on asetettu empiirisesti.
Pair-HMM on todennäköisyyksillä laajennettu FSA. Todennäköisyydet on annettu symboliparien emissioille eri tiloista, sekä niiden väliselle transitioille. Pyritään löytämään suurimman todennäköisyyden tasaus. Tähän voidaan käyttää Viterbi-algoritmia, jolla etsitään todennäköisin tilapolku. Kirjassa olevalla esimerkillä osoitettiin, että probabilistiset mallit voivat olla huonompia hauissa kuin perinteiset tasausmenetelmät, jos käytetään Viterbiä. Käytettäessä forward algoritmia voidaan päästä taas parempiin tuloksiin, jos algoritmin pisteytys saadaan riippumattomaksi yksittäisistä tasauksista.
g) Analyze the relation of the profile HMMs, HMMs and PSSMs.
Piilevät Markov mallit (HMM) ovat probabilistisia tilamalleja, joiden transitiosiirtymillä ja tiloissa esiintyvien symbolien emissioilla on annetut todennäköisyydet. Todennäköisyydet johdetaan näytejoukon avulla esim. Viterbi-algoritmilla.
PSSM:t ovat myös tilastollisia malleja, joissa symboleilla on eri todennäköisyysjakaumat eri kohdille. PSSM:n todennäköisyysjakaumat määräytyvät paikan funktiona. Toisin kuin HMM:ssä PSSM:n tilat ovat aina ketjuna, jossa transitiolla siirrytään aina seuraavaan tilaa. PSSM voidaankin katsoa olevan triviaali HMM. Tässä toteutuksessa on sarjassa identtisia tiloja joita kutsutaan match-tiloiksi ja peräkkäisten tilojen transitiotodennäköisyydet ovat 1.
Profiili HMM on samankaltainen kuin ei-probabilistinen profiili-malli ja pitkälti samankaltainen malli kuin PSSM. Toisin kuin PSSM:ssä profiili-HMM:ssä voi sekvenssin pituus muuttua. PSSM:n triviaaliin HMM toteutukseen on lisätty insert-tiloja (vastamaan välejä tasauksessa) ja silent-tiloja (jättämään pois sopimattomat kohdat tasauksesta). Profiili HMM on oleellisesti HMM-malli, joten myös tämän ryhmittelyyn voidaan soveltaa Viterbi-algoritmia.
h) Excercise 6.1 in DEKM.
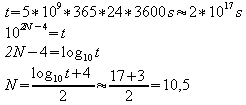
Auringon sammumiseen on viisi miljardia vuotta aikaa. Tässä ajassa ehditään tasaamaan reilut 10 sekvenssiä. Erittäin nopeaa toimintaa siis ;). Algortmi on hyvä esimerkki non-polynomial ratkeavasta ratkaisusta jollaisia ei siis todellakaan kannata käyttää.
i) Explain briefly the pros, cons, and the most useful applications of the different progressive alignment methods and profile HMMs in multiple alignment (DEKM ch.6)
j) Explain the reasons why there are methods for fragment assembly and physical mapping (S&M: ch.4 and ch.5)
Fragment assembly on tuntemattoman DNA-sekvenssin palasten kokoamista ja orientoimista oikeaan järjestykseen. Kun pyritään selvittämään tietyn DNA-molekyylin/genomin, jossa voi olla tuhansia emäspareja, koko sekvenssi, joudutaan turvautumaan fragment assembly -metodeihin. Tämä johtuu siitä, että pisimmät DNA palat joita voidaan laboratoriossa sekvensöidä ovat 700 emäsparia pitkiä, kun kromosomin pituus on 10^8 emäsparia – suora sekvensointi on siis mahdotonta. Siten pyritään saamaan useita pieniä paloja molekyylin sekvenssistä (kun esim. Käytetään shotgun -menetelmää). Näistä pienistä paloista pyritään rakentamaan fragment assembly -menetelmillä molekyylin sekvenssi käyttämällä hyväksi palasista löytyviä päällekäisyyksiä.
Kun pyritään käsittelemään tai selvittämään DNAn molekyylejä joiden koko on suhteessa kromosomiin eivät perus sekvensointi-tekniikat enää toimi. Esimerkiksi geenikartan luomiseen ei perinteisillä sekvensointi-menetelmillä pystytä pureutumaan, sillä tällöin pystytään käsittelemään pienen pieniä osia koko sekvenssistä. Physical Mapping -menetelmillä saadaan käyttöön laskennallisia menetelmiä joiden avulla voidaan käsitellä ja kartoittaa kokonaisuutta yksittäisten yksityiskohtien sijaan. Nämä menetelmät perustuvat pääsääntöisesti tiettyjen molekyyleissä sijaitsevien merkkien löytämiseen, määrittämiseen ja kokonaisuuden hahmottamiseen niiden avulla.
k) Excercise 1 in ch.4 in S&M
Molekyylin pituudeksi tulee 50 emästä, mikä on lähellä tehtävän 55 parin arviota. En käyttänyt tehtävän ratkaisemiseen muuta kuin omaa neuraaliverkkoani. Lähdin pisimmästä palasta ja pyrin sovittamaan pituusjärjestyksessä uusia paloja katsoen mitkä palat sovittuvat parhaiten (eniten sopivia emäksiä). Kolme palasta jouduin kääntämään ja komplementoimaan, jotta ne sopivat hyvin.
Sopivat järjestyksessä, f6, f1, -f4, f5, -f3, f8, -f7, f2.
f6,-f7:----------CACAGATCCGTTGAAGCCGCGGG---TACTTAACTCGAG-
f1,f2: ---------------ATCCGTTGAAGCCGCGGGC-----TTAACTCGAGG
-f4,f8:-----CCCGACACAGAT---------------GCAGTACTT---------
f5,-f3:CGACTCCCGACACA---------------CGGGCAGTACTTAA-------
12345678901234567890123456789012345678901234567890
CGACTCCCGACACAGATCCGTTGAAGCCGCGGGCAGTACTTAACTCGAGG
l) Excercise 5 in ch.5 in S&M
Explain why two restriction sites for two different restriction enzymes cannot coincide.
Restriction entsyymit leikkaavat DNA molekyylin aina tietyissä paikoissa (restriction sites), jossa esiintyy aina tietty sekvenssi. Pilkkominen tapahtuu siten että toinen säie katkeaa paikan toisesta päädystä ja toinen toisesta.
(alla kirjan esimerkki s.19)
...atccagAATTCTCGGA... ...atccag AATTCTCGGA...
...taggtcttaaGAGCCT... ...taggtcttaa GAGCCT...
Molempiin päihin jää siis tietty "sormejälki" -selvenssi, joka kannustaa katkenneen säikeen yhdistymistä saman restriction entsyymin pilkkoman toisen osasen kanssa.
Koska restriction paikan määrittävä sekvenssi on jokaiselle eri entsyymille uniikki ja katkaisu tapahtuu paikan molemmista päistä eivät paikat pysty myöskään osumaan samoille paikoille - Kertaalleen katkaistuista osista ei toinen restriction entsyymi enää löydä haluttua sekvenssiä molemmista säikeistä, koska säikeistä toinen on katkennut kesken sekvenssin. Alla olevassa kuvassa on kuvattu tilannetta jossa on kahden restriction entsyymin tunnistamat paikat "päällekäin" (GAATTC ja TTGCAA). Kun paikka on pilkottu hajoaa toisen sekvenssin emäsparien toinen säie toiseen osaan ja tunnistusta ei tapahdu ja kyseessä ei silloin myöskään ole restriction paikka.
222222 222222
111111
-----> ----->
...ATCCAGAATTCGAAGAAGAT... ...ATCCAG AATTCGAAGAAGAT...
...TAGGTCTTAAGCTTCTTCTA... ...TAGGTCTTAA GCTTCTTCTA..
<----- <- ----
r-sek1 111111
r-sek2 222222 22 2222
m) Excercise 6 in ch.6 in S&M
Lemma 6.1: A binary matrix M admits a perfect phylogeny if and only if for each pair of characters i and j the sets Oi and Oj are disjoint or one of them contains the other.
n) Excercise 8.2 in DEKM. Explain also the concept of multipilicativity here.
o) Excercise 8.7 in DEKM.
The maximum likelihood
edge lengths can be calculated in certain simple cases. Show that,
for our example of two nucleotide sequences (p. 198), the ML solution
is given by![]()
Yhtälönä on seuraava, jossa alpha merkitsee nukletodien välistä transitionopeutta.
![]()
Sijoitetaan yhtälöön u = t1 + t2 ja derivoidaan u:n suhteen ja ratkaistaan kyseisen derivaatan nollakohta. Tämä on kenties helpon tehdä Matlabissä.
%Asetetaan symbolit:
u = sym('u');
n1 = sym('n1');
n2 = sym('n2');
a = sym('a');
% ja itse funktio
P = 1/(16^(n1+n2))*(1+3*exp(-4*a*u))^n1 * (1-exp(-4*a*u))^n2;
% derivoidaan:
dPdu = simplify(diff(P, u));
% ratkaistaan Maximum likelihood dPdu:n nollakohdasta
uml = solve(dPdu, u);
% josta saadaan
% uml = -1/4*log(-1/3*(-3*n1+n2)/(n1+n2))/a
![]()
Mistä todistettavaksi tarkoitettu lause saadaan alphan arvolla 1/3. Tällöin transitionopeus on siis 1/3 merkkiä. Yritin selvittää tekstistä "mystisen" alphan valintaan liittyviä kriteereitä tai sitä mistä sen arvo johdetaan, mutta en oikein löytänyt.
EXCERCISES 2 (PART 1)
a) Too easy; smooth start: SM, Ch. 7, Ex. 13
Prove that any unoriented permutation over n elements can be sorted with at most n – 1 reversals.
Algoritmi:
Kun n = 1, on merkki varmasti järjestyksessä ilman siirtoja (n – 1 = 0, kun n=1).
Järjestetään ensimmäinen merkki paikalleen kääntämällä sekvenssi alusta ensimmäiseksi haluttuun merkkiin. Tämän jälkeen meillä on (n-1) merkkiä epäjärjestyksessä. Tätä algoritmia toistetaan taas tälle (n-1) pituiselle sekvenssille. Tästä rekursiosta voidaan laskea, että kääntöjä tarvitaan (n-1) n:n pituiselle sekvenssille.
b) Easy again: SM Ch. 8, Ex. 2
Give an example of an RNA sequence where a knot may appear.
RNA-molekyyli voidaan ajatella n:n merkin merkkijonoksi, jossa jokainen merkki kuuluu joukkoon {A,C,G,U}. Toisen asteen rakenne (secondary structure) on joukko S pareja (ri, rj) joissa 1 <= i < j <= n. Jos pari kuuluu joukkoon S vaaditaan, että ri ja rj ovat toistensa komplementteja ja j – i > t, jossa t on kynnysarvo. Kynnysarvoa tarvitaan koska RNA-molekyyli ei taitu jyrkästi eli yhtyneet kohdat eivät voi olla aivan vierekkäin.
Solmu
on taas olemassa kun
![]() ja
ja![]() ,
sekä i < k <j < l. Tällöin siis kohdat
menevät RNA-molekyylissä "lomittain".
,
sekä i < k <j < l. Tällöin siis kohdat
menevät RNA-molekyylissä "lomittain".
Esimerkiksi
ACGCUAUUUGAGU .... CCUAUGG .... CAAAAACGCA .... GGGCAUGGG
iiii kkk jjjj lll
0123 012 0123 012
0123
iiii
jossa
siis kohdikkain menevät ACGCUAUUUGAGU
...AAAC...
jjjj
0123
012
ja CCUAUGG
UAC..
210
Näiden kohtien välit kuvaavat sitä että sekvenssien väliin tarvitaan tarpeeksi tilaa, että solmu voi syntyä. Tämä pituus voi näin monimutkaisessa ja pitkissä pariryhmissä olla erittäin pitkä. Yhdistyneiden kohtien ei tarvitse olle dna-sekvenssin "kasvusuunnan" mukaan edes käänteiskomplementtejä (edeltävässä esimerkissä i ja j vastaan k ja l, katso kuva)
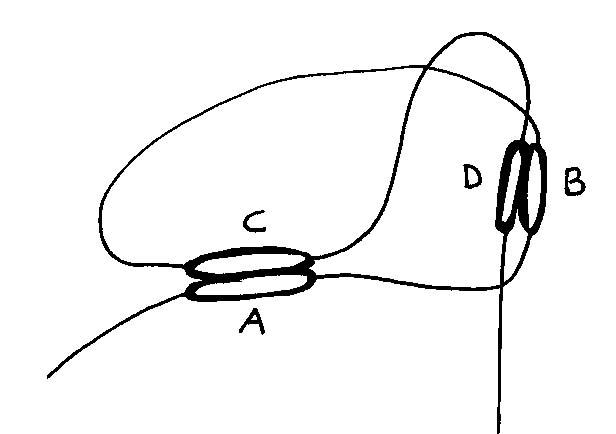
Kuvassa
on tahallaan "paskunnettu" kohdat joissa yhdistyneet
sekvenssit tai emäkset sijaitsevat, kyseisissä paikoissa ei
tapahdu mitään säikeiden jakaantumista tms. ;-).
c) DEKM: Ex 9.10
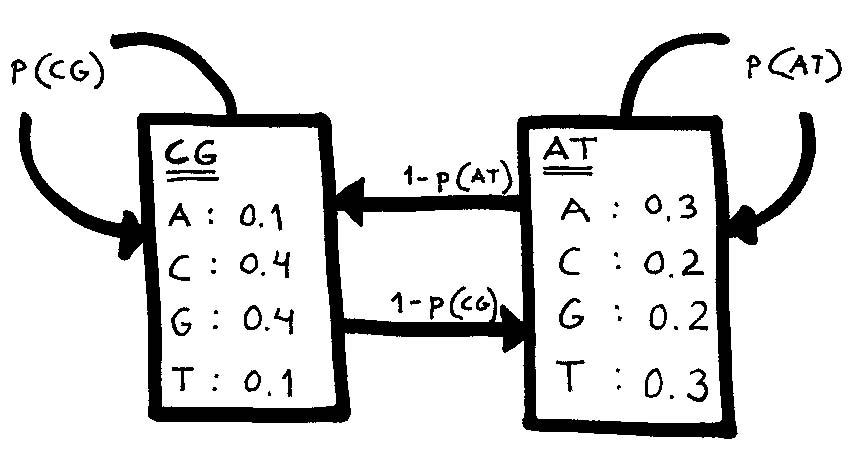
Todennäköisyyksien laskeminen:
Todennäköisyysfunktio: Jatketaan emittiota tilassa A, kunnes tulee ensimmäinen transitio tilaan B. Transitioiden lukumäärä on diskreetti satunnaismuuttuja X. Tiedetään, että transitioihin liittyvä tapahtumat ovat toisistaan riippumattomia ja jokaisen transition kohdalla transitio tilaan B tapahtuu todennäköisyydellä p, 0 < p < 1. Silloin
P(X = k) = p(1 – p)k-1 , k=1,2,...
Tämän jakauman odotusarvo saadaan seuraavasti:
![]()
Josta saadaan näppärästi todennäköisyydet p(CG) = p(S1) = 999/1000 eli tällöin juoksupituuden odotusarvo on 1000 ja p(AT) = p(S2) = 99/100 ja juoksupituuden odotusarvo = 100.
Stokastinen kielioppi: (aloitussymbolista on tehty suora siirtymä ei-terminaaliin S1, sillä tehtävässä ei puhuttu mitään aloitustilasta, voisi tehdä sitenkin että olisi yhtäsuuri todennäköisyys aloittaa kummastakin tilasta).
Aloitussymboli: S
Ei-teminaalit: {S1,S2}
Terminaalit: {a,c,g,t}
Siirtymät:
S=>S1, P=1
S1=>aS1, P=0.1*p(S1) = 0,0999
S1=>cS1, P=0.4*p(S1) = 0,3996
S1=>gS1, P=0,3996
S1=>tS1, P=0,0999
S1=>aS2, P=0.1*(1-p(S1)) = 0,0001
S1=>cS2, P=0.1*(1-p(S1)) = 0,0004
S1=>gS2, P=0,0004
S1=>tS2, P=0,0001
S2=>aS2, P=0,297
S2=>cS2, P=0,198
S2=>gS2, P=0,198
S2=>tS2, P=0,297
S2=>aS1, P=0,003
S2=>cS1, P=0,002
S2=>gS1, P=0,002
S2=>tS1, P=0,003
d) DEKM: Ex. 10.4
Nussinov-algoritmissa
pisteytykseen vaikuttaa ainoastaan delta-funktio. Kyseinen
delta-funktio (![]() )
sai arvon 1, jos i ja j muodostivat
emäsparin, ja arvon 0 jos ne eivät muodostaneet.
Tämä funktio voidaan korvata toisella pisteytysfunktiolla
)
sai arvon 1, jos i ja j muodostivat
emäsparin, ja arvon 0 jos ne eivät muodostaneet.
Tämä funktio voidaan korvata toisella pisteytysfunktiolla
![]() ,
jolla on halutut pisteytykset annetuille emäksille. Algoritmin
toimintaan tämä ei vakuta mitenkään sillä
ainoastaan pisteytykseen liittyvä funktio vaihdetaan –
muutos on erittäin helppo toteuttaa.
,
jolla on halutut pisteytykset annetuille emäksille. Algoritmin
toimintaan tämä ei vakuta mitenkään sillä
ainoastaan pisteytykseen liittyvä funktio vaihdetaan –
muutos on erittäin helppo toteuttaa.
e) SSK, Ch.11: The authors propose a method for computing a substituting matrix for amino acids. Characterize the differences from the PAM matrices. Comment the proposal. Where would you use each?
f) Explain SSK, Ch. 12 eqn(2) (p.242) Use at most 1 page.
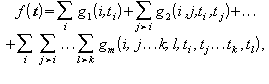
g) Paper by Chen et al (regulatory networks): try to justify the Maximum gene regulation problem.
h) Paper by Tamayo et al: "Translate" the SOM algorithm to the more familiar notation (see, e.g., http://www.cis.hut.fi/research/som-research/som.html)